
| Home | Syllabus | Schedule | Lecture Notes | Extras | Glossary |
| Home | Syllabus | Schedule | Lecture Notes | Extras | Glossary |
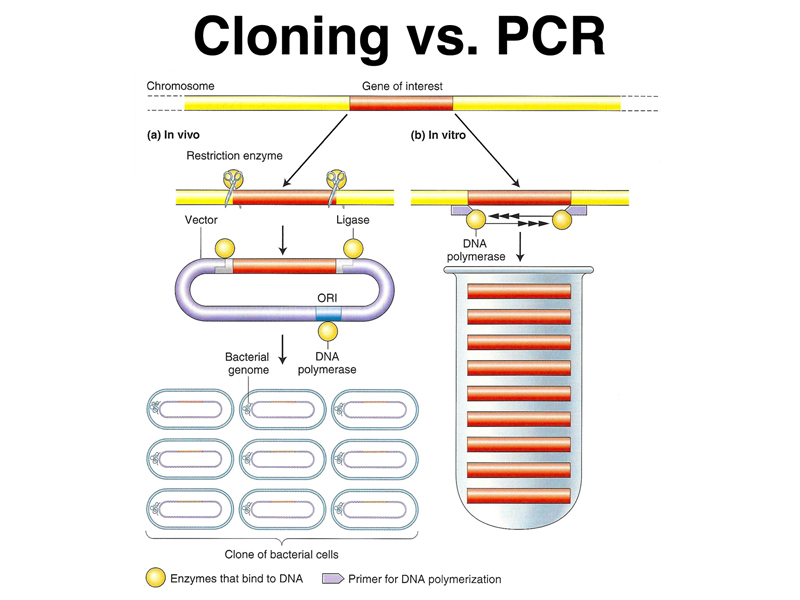
We began by reviewing our introduction to recombinant DNA technology in the last lecture. To study genes, we need to be able to isolate genes in quantity. There are two kinds of methods, as shown below. The Polymerase Chain Reaction or PCR is an in vitro method of amplifying a specific DNA sequence specified by two flanking primers. In order to amplify a piece of DNA, we must know the sequences at the ends in order to be able to synthesize the primers. Molecular cloning is a collection of methods that allow single DNA (or cDNA copies of RNA) molecules to be inserted into a variety of different types of cloning vectors that can be grown in bacteria. Molecular cloning does not require knowledge of the sequence to be cloned, and relies heavily on clever screening methods to identify the molecule of interest.
The figure below shows a few kinds of cloning vectors that differ in their capacity to accept foreign DNA. Plasmids, adapted from bacterial drug resistance plasmids, can accept insert of up to 5 kb in size, while engineered phage λ can accept insert up to 15 kb in size. Cosmids and other engineered vectors (fosmids, BACs, etc.) can accept much larger inserts, up to 200 kb.

The key to molecular cloning was the discovery of restriction enzymes, endonucleases that cut DNA at specific recognition sites. The figure below shows the cut made by EcoRI. Because the ends of DNA molecules cleaved by EcoRI are "sticky," two different DNA molecules with identical sticky ends can be ligated together to make a recombinant DNA molecule. The two different DNA molecules can be from any source, even widely separated species. There are hundreds of restriction enzymes with different recognition sites.

Once we understand cloning vectors, restriction enzymes, and ligation, we are ready to construct genomic libraries, as shown below. An entire genome can be cleaved into thousands of different restriction fragments and cloned into a cloning vector.

Transformation of E. coli with such a collection of molecules will produce bacterial colonies that each contain a different piece of the genome. The entire collection of clones is called a genomic library, although the collection is not organized the way that the books in a library are organized, and we use clever screening techniques to identify the clones of interest.

Cloning vectors need a bacterial origin of replication, a selectable marker, and a convenient multiple cloning site that has recognition sites for a set of restriction enzymes. The plasmid shown below contains the multiple cloning site in the lacZ gene so that plasmids that have ligated back together without accepting an insert can be recognized as lacZ+ using X-gal.

We also introduced the subject of biotechnology. One example, shown below, is the production of human insulin by engineered bacterial cells. A human cDNA clone is expressed in bacteria under the control of a bacterial promoter. Because the genetic code is universal, human insulin is synthesized on bacterial ribosomes, and can be produced in quantity in fermentation tanks. There are currently quite a number of human proteins on the market as drugs (human growth hormone, EPO, etc.) that are manufactured in this way.

There are many research and commercial applications for genetically engineered organisms beyond bacteria. There are many ways to introduce foreign DNA into cells, as outlined below.

Transformation. This is the simplest method. It is commonly used to introduce DNA into bacterial cells; it also works for yeast. Cells are prepared in some way to make them more able to take up DNA, and are simply mixed with pure DNA. This is not a very efficient process, making selectable markers on the cloning vectors very important.
Viruses. We were introduced to viruses as vectors when we reviewed engineered types of bacteriophage λ that are used to transform E. coli. In response to a question from a student about whether there were any transgenic humans, the answer is yes in a very limited way. A small number of people have been treated with somatic gene therapy to correct a genetic defect. The changes to their genome are not heritable. Early gene therapy was carried out with a viral vector, but the death of a patient a decade ago has made it necessary to find other methods, which are currently emerging.
Injection. Microinjection with a very fine glass needle is the method used to transform Drosophila, as we shall see. It is useful in transforming animal cells, which lack a tough cell wall.
Projectile Gun. The tough cell walls of plant cells are best dealt with using this extremely cool method. The DNA to be introduced into plant cells is used to coat a number of microscopic particles of tungsten. The DNA-coated tungsten particles are dusted onto an actual bullet. The bullet is fired at a plate with a hole in it just smaller than the bullet, above an open dish containing plant cells. As the bullet is stopped by the plate, the tungsten particles continue toward the plant cells at the speed of the bullet. They penetrate the plant cells, carrying their DNA. Subsequent versions of this technique use air pressure and plates that fracture at specific pressure levels, taking much of the fun out of the technique while slightly improving the results.
Yeast is a unicellular eukaryote that has been used for thousands of years to produce bread, beer, and wine. The long human experience with culturing yeast makes it a great organism for the industrial production of proteins that might require eukaryotic post-translational modifications. Yeast is also an important model organism for basic research. Yeast is easily transformed in culture with pure DNA. Several different kinds of yeast vectors are shown below.

Yeast integrative plasmid. This type of yeast vector has a bacterial origin of replication and a bacterial selective marker (e.g. ampicillin resistance) to allow it to be grown in E. coli. It lacks a yeast origin of replication, but carries a yeast selectable marker, such as an gene for an enzyme required for the biosynthesis of an amino acid. There is a multiple cloning site to accept DNA inserts. When yeast cells are transformed with this plasmid, recombination between the plasmid and the yeast genome at the site of the selectable marker integrates the entire plasmid into the yeast genome.
Yeast episomal plasmid. This type of yeast vector exists as an episome at a high copy number per cell, and works just like a bacterial plasmid.
Yeast centromere plasmid. This is a circular plasmid containing a cloned yeast centromere. Centromere plasmids are maintained at one copy per cell, just like small circular chromosomes.
Yeast artifical chromosome. The addition of yeast telomeres to a centromere-containing plasmid creates a yeast artificial chromosome, capable of accepting very large inserts.
Drosophila continues to be a key model organism. The introduction of methods to transform Drosophila in the late 1980s was a very important development. The image below shows transgenic Drosophila third-instar larvae expressing Green Fluorescent Protein (GFP, from a jellyfish) in their salivary glands.

Transformation of Drosophila was accomplished by engineering a mobile genetic element called the P element. P elements are a class of DNA transposons that have two important parts: a gene that encodes a transposase that catalyzes the transposition of the P element, and special sequences at the ends of the P element that are recognized by transposase. P elements can insert at thousands of different sites in the Drosophila genome. Old laboratory strains of Drosophila that are derived from flies caught in the wild before 1950 lack P elements, while most current wild populations of Drosophila melanogaster carry P elements. Apparently there was a horizontal transfer from another species some time in the mid-20th century.
As shown below, a P element that has has a partial deletion of the transposase gene becomes nonautonomous: it does not encode functional transposase, so it can no longer catalyze its own transposition, but it still be mobilized using transposase from another functional copy of the P element.

The P element has been engineered into two types of elements to carry out Drosophila transformation. The first type is the helper element "wings clipped," which encodes functional transposase. The second type is a series of elements that have functional ends but no functional transposase. The transposase gene has been replaced by genes for morphological markers (like the familiar white gene) and a multiple cloning site that allows other DNA to be inserted.

As shown below, to transform Drosophila, embryos from a strain bearing the appropriate markers (e.g. w, if w+ is being used to mark the P element) are injected with a mixture of the engineered P element and the "wings clipped" helper plasmid. The site of the injection is the posterior pole of the embryo, the site of formation of pole cells, the future germ line.

As shown below, the P element transposase mobilizes the engineered P element from the plasmid, where it is free to integrate into many different chromosomal sites.

The figure below gives an overview of the transformation process. Embryos injected with the plasmid mixture develop into adults that bear insertions of the element in some cells of their germ line. Note that the transformed adults still have white eyes, as they are chimeras, with only a few cells in their germ line carrying the w+ allele. These are bred to other white-eyed flies. Any red-eyed offspring are transformants carrying the w+ allele as well as any other DNA that was in the engineered P element.

Transgenic plants are of enormous economic importance in the USA, with the vast majority of the American corn and soybean crops being transgenic. Transgenic crop plants are potentially of tremendous value at improving the nutritional qualities of crop plants, as well as improving yield.
One method of making transgenic plants uses Agrobacterium tumefaciens. This is the bacterium reponsible for crown gall, tumorous growths seen on a wide variety of plant species in reponse to infection by virulent strains of Agrobacterium. As shown below, Agrobacterium has a virulence plasmid (Ti plasmid) with a mobile genetic element called T-DNA. The T-DNA leaves the plasmid and integrates into the genome of the host plant, where it causes uncontrolled cell growth as well as directing the plant to synthesize compounds that are used by Agrobacterium as a food source.

The image below shows how Agrobacterium multiply in the crown gall itself after transforming the host.

As shown below, the tumor-inducing genes can be removed from the Ti plasmid and replaced with genes to be introduced into the plant genome. In this example, the plasmid is modified to transform the host with genes required for β-carotene (provitamin A) synthesis.

The image below shows the steps necessary to make a transgenic plant. In this example, tobacco is used as the host. We all enjoyed the irony of being able to give cancer to tobacco.

Transformed plants (always heterozygous for the insertion) can be selfed to produce the familiar 1:2:1 ratio of types of offspring, including the desired true-breeding homozygotes.

The transgenic plant from the example in your text is Golden Rice, shown below. Rice is the major source of calories for a substantial fraction of the world's population. Unfortunately, rice lacks vitamin A (or rather provitamin A), so many of the world's poor suffer from vitamin A deficiency. The first symptoms are night blindness, but full blindness can also develop. Golden Rice was developed as a way of addressing vitamin A deficiency in poor populations that rely on rice as a major food source.

Unfortunately, Goden Rice has been widely rejected by the governments of countries containing most of the people who would benefit from it, largely for cultural and symbolic reasons that I am not embarrassed to characterize as at best recklessly irresponsible. Following my brief but intense sermon on the subject in class, a student asked whether there had also been genetic modification of corn (maize).
The development of maize is, in my view, the single greatest achievement of agricultural genetics in human history. The wild ancestor of maize, called teosinte, is an unexceptional plant that requires a great deal of imagination to recognize as a food source. Selective breeding over thousands of years produced the amazing crop that is modern maize. A student sought to characterize modern transgenic maize as having been produced by artificial means. In response, I pointed out that there was nothing natural about the thousands of years of selective breeding that produced modern maize, which is one of the most unnatural plants that I know. Consider the mechanism of seed dispersal of maize: the domestication of humans, who extract maize kernels from the ripe ears and plant them in rows.
The laboratory mouse is the model organism most closely related to humans, and is the workhorse of biomedical research most directly applicable to humans. The gene set of humans and mice are essentially identical. Transgenic mice are important models for human diseases with a genetic component. Two of the three mice shown below are transgenic, carrying the gene for Green Fluorescent Protein (GFP).

While foreign DNA of any type can be introduced into mice, the production of knockout mice is a particularly useful technique. Throughout much of this course, we have talked about the genetic investigation of many processes. Biochemical pathways were first understood by obtaining mutations that blocked specific steps of the pathway, leading to the idea that each gene encoded a specific enzyme. We understood genes controlling development in Drosophila by obtaining mutations that caused defects in development. These kinds of approaches are now called forward genetics, which we can define as the collection and study of mutations that cause a specific phenotype.
The relatively modern approach of reverse genetics seeks to discover the function of a gene by producing targeted mutations in that gene. This requires a cloned copy of the gene, which is then engineered to be defective, and is used to destroy the function of one of the endogenous copies of the gene in the normal genome.
Targeted mutations in the mouse that destroy the function of a gene are called knockout mutations or simply knockouts. The steps necessary to produce a mouse knockout are outlined below.
In the figure below, a clone of part of a mouse gene with two exons is shown. This part of the gene is cloned into a targeting vector containing the thymidine kinase gene (a selectable marker). Another selectable marker, the gene for neomycin resistance, is inserted into Exon 2 of the mouse gene. The circular plasmid is linearized by cutting at a unique restriction site. The DNA is used to transform mouse embryonic stem cells in culture. Embryonic stem cells are capable of participating in development to produce any part of a mouse.

Once the linearized DNA is introduced into an ES cell by transformation, three things are possible, as shown in the figure below. First, Exon 2 might recombine with one of the chromosomal copies of the mouse gene to replace the chromosomal copy of Exon 2 with the engineered copy that has ben interrupted by the neomycin resistance gene. This event will not incorporate the thymidine kinase gene into the chromosome. Second, the entire targeting construct might recircularize and integrate elsewhere in the mouse genome. This event will incorporate both thymidine kinase and neomycin resistance into the genome of the ES cell. Finally, the DNA introduced by transformation might not integrate anywhere in the genome, and both markers will be lost.
As shown in the bottom part of the figure, we then add two drugs to the culture: one kills all cells that lack neomycin resistance, while the other kills all cells that have a thymidine kinase gene. This selects for the first event, recombination of the targeting construct with the chromosomal copy of the gene. Cells that survive the selection are then analyzed by PCR to confirm the targeted mutation.

Once the genotype of the ES cells has been confirmed, they are injected into blastocyst-stage embryos from a strain with a different coat color than that used to produce the ES cells. The chimeric embryos are then implanted into a pseudopregnant surrogate mother, where they complete development. Chimeric mice are recognized among the offspring by their patches of different coat color.

Chimeric mice are bred to an inbred strain to recover gametes derived from the ES cells as shown below. Once mice heterozygous for the targeted allele are obtained, they are bred together to attempt to produce mice homozygous for the knockout. The figure shows these as having an easily recognizable phenotype (curled tail), but it is also possible that the mice homozygous for the knockout will be phenotypically normal, or that they will fail to complete development. If they fail to complete development, developing embryos from the mating of two mice heterozygous for the knockout can be studied to determine what aspect of development is failing.

The figure below shows a chimeric mother and two of her pups. The chimera exhibits two different eye colors and two different coat colors.

There is currently an ambitious project, the Knockout Mouse Project (KOMP), with the goal of producing a knockout allele of every mouse gene. The project currently has obtained ES cells with knockout alleles of 14,000 single genes, and has the goal of recovering mice from 5,000 of these ES cell lines by 2016. Each knockout will be subjected to detailed phenotypic analysis. The project will eventually catalog the phenotype of knockouts of every gene in the mouse genome. This is expected to give us valuable insights into the function of the corresponding human genes. The precise function of a substantial fraction of the 22,000 genes in the human genome is unknown.
We would like to be able to identify the genes responsible for inherited disorders in humans. Isolation of disease genes by molecular cloning offers, at a mimimum, the ability to identify carriers in human families. Eventually, we hope to develop new treatments for inherited disorders by understanding the function of the genes associated with the disorder. As an example, we will use the gene responsible for Huntington Disease, an autosomal dominant neurodegenerative disorder that we discussed early in the course.
We begin with a review of the basics from the last lecture. Any cloned segment of the human genome can be used to identify the same sequence on filters using nucleic acid hybridization. Denatured probe and denatured target DNA are incubated under carefully chosen temperature and salt conditions to allow only the chosen sequences to hybridize. The sequences hybridizing to the probe can be identified by autoradiography if a radioactive label is used, or by a chromogenic or luminescent chemical reaction, if a chemical label is used.

The figure below shows hybridization of a labeled probe in more detail. While many sequences will match short segments of the probe, we pick conditions that require an exact match over a substantial portion of the sequence.

We have previously discussed agarose gel electrophoresis of DNA. The figure below shows a gel box for horizontal electrophoresis, in which negatively-charged DNA migrates to the postive electrode through the gel. Smaller fragments move more quickly.

Just as we can produce a replica of a plate bearing bacterial colonies or phage plaques on a filter, we can produce a filter that is a replica of an agarose gel. The gel is treated with chemicals that nick DNA, then denature it, allowing denatured DNA to be transferred to a filter by capillary action. This technique is called Southern blotting (the last name of the inventor is Southern). There are two subsequently-invented techniques with similar names. If RNA rather than DNA is run on the gel before transfer, it is called a Northern blot. If proteins are run on a gel and then transferred for antibody detection, it is called a Western blot.

After a couple of hours to allow transfer, we can remove the filter replica of the gel for further processing.

The filter is incubated in a plastic bag with labeled probe at the appropriate temperature.

After being washed to remove unbound probe, the filter is read for detection.

In this example, a radioactive probe was used, allowing detection by autoradiography.

The figure below shows the entire process. Note that if we use a restriction digest of a complete genome, we will see only a smear rather than individual bands, but individual fragments will still sort by size.

Many of the restriction sites in the human genome are polymorphic, with two alleles present in the population. The figure below illustrates a polymorphism for an EcoRI site. In the allele on the left, the site is present. In the allele on the right, one of the bases of the EcoRI site is a variant that eliminates the restriction site. This means that the size of the restriction fragments that hybridize to a cloned probe covering this site will be different for the two alleles, which is why this kind of variation is called a restriction fragment length polymorphism or RFLP.

In a EcoRI digest of genomic DNA, the two alleles will produce fragments of different sizes that are detected by a probe, as shown below.

As shown below, homozygotes for either allele will have single bands hybridizing to a probe, while heterozygotes will show both bands. Genetically, RFLPs behave as codominant alleles. Because about 98% of the human genome is noncoding, most RFLPs will be selectively neutral. Even RFLPs in coding regions can be neutral if the base substitution is a synonymous mutation.

Using Southern blots and cloned segments of the human genome to identify RFLPs gives us an unlimited supply of selectively neutral variants across the entire human genome. This allows us to search for RFLPs that show linkage (the failure of independent assortment) with disease genes.
Researchers studying Huntington Disease found a small village in Venzuela where a European founder carrying the variant allele for Huntington Disease had fathered children. There was an extensive pedigree showing the inheritance of the disoder, shown below. Because the disease was introduced by a single founder, all affected individuals carry the same risk allele. Recombination is infrequent enough at a small scale in the human genome that any genetic variation closely linked to the risk allele should be the same across the entire pedigree.

There was a similar pedigree of affected individuals in the USA, shown below.

The researchers obtained genomic DNA from individuals in these large pedigrees and began to use random cloned segments of human chromosome 4 (where the Huntington Disease risk allele was known to be located) to identify RFLPs. Each RFLP was tested for linkage to Huntington Disease. Results for one probe, called G8, are shown below.

Notice that a single band at 4.9 kb is polymorphic. Some individuals have this band, others do not. The presence (+) or absence (-) of this band is marked in the image below. If you have good eyes, you can see a polymorphism for another site, which shows up as a 17.5 kb or 15 kb band.

A map of the restriction sites identified by probe G8 is shown below.

Of many probes tested, probe G8 showed tight linkage to Huntington Disease (HD). The table below
shows lod scores for the hypothesis of no recombination (θ = 0) between the RFLPs identified
between G8 and the Huntington Disease risk allele. Combining the pedigrees gives a lod score of
greater than 8, meaning that it is 10 The discovery of linkage of HD to probe G8 allowed a small region of the human genome to
be examined one gene at a time to find the gene responsible for Huntington Disease. The responsible
gene was named huntingtin (HTT). Variant alleles of HTT that cause disease have an abnormal
number of glutamine residues in a specific portion of the encoded protein. The polyglutamine
tract expands like the VNTR or STR loci that we discussed when reviewing forensic PCR.
As shown below, there is variation for the number of glutamine residues (CAG repeats) in HTT.
People who develop Huntington Disease have 40 - 60 CAG repeats in the relevant part of the
protein. People who do not develop Huntington Disease have fewer than 35 CAG repeats. Intermediate
numbers are characterized as "premutation" alleles.
The CAG repeats in the HTT gene can be analyzed by PCR, using primers to the part of the
gene flanking the CAG repeats. The image below shows the PCR products from lymphocytes (L) or sperm (S) from three
individuals with risk alleles. The number of repeats is somatically unstable, producing the multiple
bands shown.
This means that there is now a diagnostic test for individuals known to be at risk for Huntington Disease.
Half of the children of affected carriers are at risk. Prior to this work, they had to wait until
their fifth decade to discover if they were going to develop Huntington Disease. Now, anyone with
a family history can discover their status at any age with a simple genetic test.
Earlier in the semester, five students won genetic testing from 23andMe in an essay contest. They
donated DNA by collecting and processing their saliva using a sample kit provided by the company. A
person using a "spit kit" is shown below.
Samples are analyzed for 600,000 single nucleotide polymorphisms (SNPs) using an Affymetrix SNP chip, shown below.
The GeneChip is a device built using the same technology that is used to produce integrated circuits.
It takes advantage of the complete sequencing of the human genome and of information derived from
surveys of human populations. Each GeneChip has millions of spots, each containing a different single-stranded
oligonucleotide that contains a site of variation. For each such site, the four oligonucleotides containing
each of the four possible bases at that site are adjacent to each other on the chip.
As shown below, hybridization conditions are chosen that will not permit binding of DNA if there is
a single-base mismatch.
As shown below, genomic DNA from a sample is prepared by shearing it into small pieces,
melting it to single strands, and modifying each molecule by adding a biotin tag.
As shown below, hybridization of a biotin-tagged fragment of genomic DNA to a particular spot on the chip allows
that spot to be bound by a molecule of the biotin-binding protein streptavidin, which for this
application is fluorescently tagged.
As shown below, at most two of the four similar oligonucleotides will bind to genomic DNA from
a particular individual (because they can have, at most, two alleles). The vast array of spots
is scanned, photographed, and scored by a computer. The image on the right below shows a photograph of
a chip ready to be scored for a very large number of SNPs.
This technique shows the enormous progess in the analysis of the human genome from the early days
of scoring RFLPs using Southern blots in 1983 (the year that the G8 probe was found to be linked to HD)
to the consumer genomics of 2013. We will later explore how the data from a GeneChip is
used to predict an individual's risk of various diseases.





SNP chips





