
| Home | Syllabus | Schedule | Lecture Notes | Extras | Glossary |
| Home | Syllabus | Schedule | Lecture Notes | Extras | Glossary |
We began by reminding everyone that now that the third exam is over, the raw material for much of the final is available for study. Please see the answer keys for Exam 1, Exam 2, and Exam 3. Questions that a substantial portion of students answered incorrectly will be reworked for the Final Exam, scheduled for Thursday, December 12.
We began our discussion of recombinant DNA with a statement of our goals. If we are ever to have any hope of understanding the nature of a specific gene, we have to be able to work with that gene in pure form. This means that we need techniques that will let us produce many identical copies of a specific gene. We were already introduced to the concept of amplifying a segment of DNA using the polymerase chain reaction (PCR). Please see the introduction to PCR and the description of the use of PCR for forensic DNA analysis in the previous lecture notes.
We asked whether anyone saw a potential bottleneck in the use of PCR, and a student correctly pointed out that in order to amplify a particular DNA sequence, we had to know the base sequence of at least the ends of the sequence to be amplified, in order to be able to synthesize PCR primers. This raises the question of what sort of techniques were used to isolate genes and to determine their sequence before there was PCR. The techniques of molecular cloning predate PCR. The two techniques are outlined in the figure below.
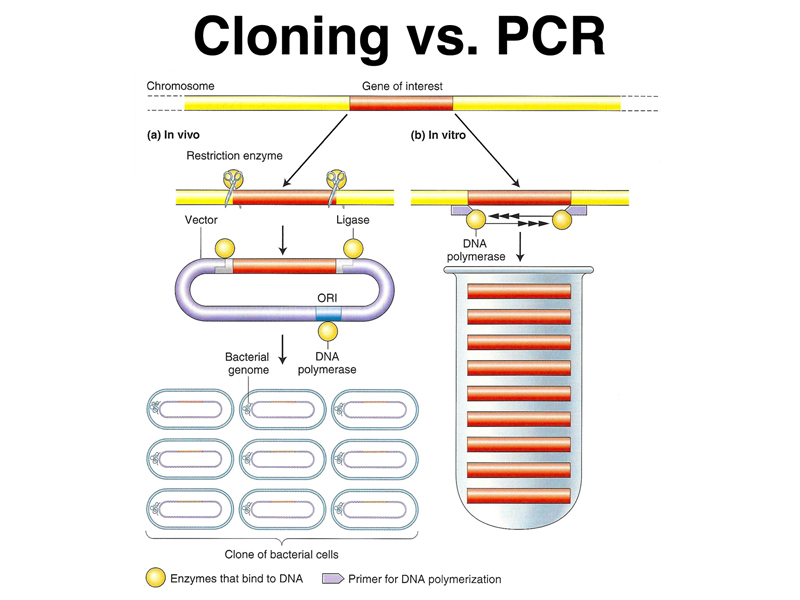
In brief, as shown on the left, in molecular cloning, genomic DNA is fragmented and incorporated into vectors that allow the sequence to be propagated in E. coli. Growth of a bacterial cell transformed with a single molecule of DNA allows that sequence to be amplified as needed. In contrast, in PCR, the sequence of interest is amplified in vitro using many rounds of replication and specific primers.
We want to make clear at the outset that cloning has two senses in modern biology: molecular cloning, the production of many copies of a single DNA molecule through growth in bacteria, and organismal cloning, the production of genetically identical animals through artificial asexual reproduction, beginning with Dolly the sheep in the late 1990s.
We would like to be able to clone DNA molecules to analyze the structure and function of specific gene products. Over the course of this semester's lectures, we have heard many stories of specific human genes that cause hereditary disorders. In most cases, we didn't know the precise nature of the gene product that causes a particular hereditary disease until the gene was isolated and sequenced.
Another application of molecular cloning is biotechnology. Once we are able to isolate specific genes and manipulate them, it becomes possible to turn genetics into a branch of chemical engineering where specific proteins with an industrial or medical use can be produced in large quantities. We will briefly examine a few examples of this process today.
We then began a checklist of everything that we need to carry out molecular cloning. First, we need DNA, which is very easy to obtain. Members of the class who had their genome analyzed by 23andMe earlier in the semester simply collected some of their saliva in a sample tube, mixed it with a few simple chemicals built into the sample collection device, and dropped the sample into the mail. Genomic DNA is readily obtained from cells using very simple chemistry, which is the same for all DNA. Some members of the class have isolated DNA from strawberries using dish detergent and isopropyl alcohol. Students agreed that obtaining DNA was not a problem.
Sometimes, we would like to convert mRNA into DNA for molecular cloning. We can use viral reverse transcriptase, an enzyme that allows RNA viruses to replicate, for this purpose. Reverse transcriptase, or RNA-directed DNA polymerase, makes a DNA copy of an RNA molecule. The enzyme requires a primer; because eukaryotic mRNAs have a poly-A tail, we can use oligo-dT as the primer. The steps required to turn a collection of RNA molecules into double-stranded DNA are shown in the figure below. RNA, but not DNA, is destroyed by exposure to high pH (NaOH), so after the RNA strand is copied into DNA, we can destroy the RNA and replicate the other strand with DNA polymerase as shown.

It is very helpful to be able to make cDNA copies of eukaryotic mRNAs, because for some genes, the vast majority of the bases in the genomic copy of the gene are part of introns. It is fairly difficult to predict the intron/exon boundaries in genomic DNA, making cDNA copies of mature mRNA very important in the analysis of a specific gene.
In order to propagate individual molecules of genomic DNA or cDNA we need vectors. These are adapted from the plasmids and bacteriophages that we discussed in our review of prokayotic genetics, as shown below.

We previously discussed multidrug resistance plasmids that arose shortly after antibiotics were first used to fight bacterial infections. These plasmids replicate independently of the bacterial chromosome and carry genes that confer resistance to antibiotics. Some of the first cloning vectors were adapted from such plasmids. The figure below illustrates the concept of introducing additional DNA into a plasmid. Bacteriophages, specifically λ, have been adapted into cloning vectors as well. Another kind of vector is the cosmid, built from the cohesive end sites of phage λ. The vectors differ in their cloning capacity. Plasmids can take up to about 5 kb of DNA, phage λ vectors can take 15 kb, and cosmids can take 40 kb. Not shown are some newer vectors such as fosmids, derived from the bacterial F factor, with a capacity of 50 kb, and bacterial artificial chromosomes or BACs, with a capacity of 200 kb. All of these vectors can replicate in E. coli.

Now that we know that we can obtain DNA of interest and that we have cloning vectors, we need a few more bits of technology to complete our molecular cloning kit. The next essential component is restriction enzymes.
Restriction enzymes were discovered as bacterial restriction-modification systems that protect bacteria from phage infection. Students agreed that it would be a good idea for bacteria to be able to recognize and destroy foreign DNA in their cells to protect against bacteriophages. The first systems studied had two components: a modification enzyme that recognizes a specific short DNA sequence and methylates it, and a restriction enzyme that recognizes the same specific short DNA sequence and cuts both strands if the sequence is not methylated. The bacterial chromosome is protected from the restriction enzyme by the modification enzyme, but any unmodified DNA that enters the cell will be cleaved by the restriction enzyme.
The action of one of the first restriction enzymes studied, EcoRI, is shown in the figure below. EcoRI recognizes a palindromic sequence and makes a staggered cut. Notice that the ends generated by EcoRI cleavage are "sticky," that is, any two DNA molecules that have ends generated by EcoRI cleavage can anneal by complementary base pairing. The nicks can be sealed with DNA ligase.

The figure below combines all of these ideas to show the generation of a recombinant DNA plasmid carrying a foreign DNA molecule. Both the plasmid, which has a single EcoRI site, and genomic DNA of interest are cleaved with EcoRI. The mixture of cleaved plasmid and genomic DNA is incubated in the presence of ligase to generate a library of plasmids, each containing an individual DNA molecule.

A collection of such recombinant DNA molecules can be introduced into E. coli cells by transformation, as shown below. Each transformant has received a different molecule. The thousands of different pieces of a genome, each recovered in a different plasmid and introduced into a bacterial cell, constitute a library of genomic sequences. We can use different restriction enzymes to make a set of libraries in order to ensure that all of the sequences in the genome are represented.

We need to make sure that we can distinguish plasmids that have been ligated back together without picking up a genomic insert. Plasmid vectors have been engineered to allow this. The figure below shows a plasmid vector that has an origin of replication, a selectable marker (ampicillin resistance), and a multiple cloning site (a sequence with recognition sites for multiple restriction enzymes) in the middle of the lacZ gene. Plasmids that have not picked up an insert will have a functional lacZ gene, while recombinant plasmids that have incorporated a piece of DNA will be lacZ-.

The figure below shows a plate with colonies that have been transformed with a mixture of recombinant plasmids. The plate contains ampicillin to kill all bacterial cells that have not picked up a plasmid, and also IPTG and X-gal. The transformed cells are chromosomally lacZ-. Cells that have received a plasmid without an insert are lacZ+ and blue, while cells that have received a plasmid with an insert are lacZ- and white.

Next, we need a way to detect bacterial cells that contain a DNA sequence of interest. One way to detect these cells is through nucleic acid hybridization. As shown below, if we create a nucleic acid probe labeled with radioisotope or a chemical tag, we can detect DNA that has the complementary sequence by denaturing the DNA and hybridizing it to the probe. We can vary the temperature and salt concentration to adjust the stringency of the hybridization reaction, creating conditions that require a perfect match or that allow some mismatches of the probe.

We can carry out hybridization of a labeled probe to DNA picked up on filters in a process similar to replica plating. First, we plate the library on a series of numbered plates. Each plate will contain a large number of colonies, each with a different recombinant plasmid containing a different part of the genome. We make a filter replica of each plate as shown below.

We treat each filter with a solution containing detergent to lyse the bacteria and NaOH to denature the DNA, as shown below. Each colony will produce a spot of DNA on the filter. The DNA spot will contain the entire chromosome of the bacterium as well as recombinant plasmid DNA. We then hybridize each filter to the labeled probe.

DNA spots that have hybridized to the labeled probe can then be detected using X-ray film (for radioactively labeled probes) or a chemical reaction (for chemically labeled probes). We then go back to the original plate to recover the colony containing the plasmid that hybridized to the probe.

The strategy of using a nucleic acid probe to identify plasmids of interest depends on being able to create a probe. We discussed a few examples of such strategies. Suppose that we know the sequence of the protein product of a gene of interest, for example the growth hormone used as the example in your text. The techniques for sequencing proteins were developed before the techniques for sequencing DNA. We can use our knowledge of the genetic code to generate a set of all possible DNA sequences that might encode a particular series of amino acids. There are computer programs that will search a protein sequence for a region of minimal degeneracy to reduce the number of sequences that we need to synthesize.
Another example of a nucleic acid probe would be to use a cDNA clone to recover the genomic sequences for the gene encoding that transcript. We might also use a genomic probe to recover cDNA clones from a cDNA library.
In our discussion of developmental genetics, we described the conservation of homeotic genes from Drosophila to humans. Once the Drosophila genes were cloned, they were used as probes to screen libraries of human or mouse genomic DNA at reduced stringency to recover clones of the mammalian orthologs of homeotic genes.
There is another strategy for recovering the coding sequences for a protein of interest if we have an antibody to that protein. This requires the construction of a special cDNA library in an expression vector, as shown below.

Once we have made cDNA from the cell type of interest, we sonicate the cDNA to shear it into small pieces, adding EcoRI linker sequences to both ends of each fragment. These fragments are then cloned into an expression vector using an EcoRI site in the lacZ gene. Each recombinant phage will express a fusion protein consisting of the β-galactosidase and a short amino acid sequence encoded by the insert.
Using our antibody probe, we can screen the expression vector library using replica filters as before, as shown below. In this case, the filters retain a spot of proteins that represent all of the proteins present in the plaque, including the fusion protein.

We use our antibody and a secondary antibody labeled with radioisotope or a chemical tag to identify plaques expressing a protein that reacts with our antibody, as shown below.

Now that we have introduced the subject of recombinant DNA, we will briefly introduce another application of recombinant DNA techniques: biotechnology.
There are some human disease conditions that require human proteins as treatment. The most familiar example is treating diabetes with insulin. Insulin is a peptide hormone. It can be obtained from pigs, but the human insulin gene was isolated some time ago, making it possible to create bacteria that manufacture human insulin, as shown below. A cDNA clone of the human insulin-producing gene is built into a recombinant bacterial plasmid that expresses the cloned sequence under the control of a bacterial promoter. The genetic code is the same in E. coli and humans, so the mRNA from the human insulin gene is transcribed and translated. The genetically engineered bacteria can be grown in a large fermentation tank, and human insulin can be recovered from the culture.

Other examples of currently approved human proteins from recombinant bacteria include human growth hormone (for the treatment of pituitary dwarfism) and erythropoetin or EPO (for the treatment of some specific types of anemia).
DNA sequencing is an important part of the analysis of genes that have been isolated for the study of their structure and function. Here we will review a specific technique for DNA sequencing, Sanger sequencing, which was one of the first methods developed and which is still in use today.
Sanger sequencing is based on using modified nucleotides. The principle modification is that the special nucleotides required for Sanger sequencing are dideoxyribonucleotides rather than deoxyribonucleotides. The structure of deoxyribose and dideoxyribose are compared below. Note the absence of the hydroxyl group on the 3' carbon.

Remember that the 3' hydroxyl group will be part of the sugar-phosphate backbone.

The consequences of incorporating a dideoxyribonucleotide into a growing DNA strand are shown below. Because the dideoxyribonucleotide lacks a 3' hydroxyl, it cannot form a phosphodiester bond with the next nucleotide. Therefore, dideoxyribonucleotides will act as chain terminators, preventing any additional elongation of that DNA strand as shown below.

Imagine that we have a primer-template mix ready for elongation by DNA polymerase, as shown below.

Imagine that we set up four reactions with the primer-template mix. Each reaction will have the template, the primer, DNA polymerase, and all four dNTPs, as shown in the figure below.

Before we start the synthesis of the new strand, however, we will poison each reaction with a small quantity of a single dideoxyribonucleotide, as shown below. In the early days of Sanger sequencing, the dNTPs were radioactively labeled, but beginning in the 1990s a different color fluorescent dye was attached to each ddNTP. Imagine that we are using fluorescence.

As shown below, as the new strand is synthesized in the reaction poisoned with ddATP, every time an A is incorporated, it might be dATP or it might be ddATP. If a ddATP is incorporated, synthesis of the new strand will terminate. This will produce a collection of DNA strands all ending in A. The reactions poisoned with the other three ddNTPs will produce similar products.

Because we are using a primer to start the synthesis of the new strand, every newly-synthesized strand will start at exactly the same position (the 5' end of the primer). The four reactions will produce a set of products that differ only in the 3' end, with each new strand ending with the incorporation of a chain-terminating ddNTP. As shown below, if we run the reaction products in adjacent lanes of a gel that sorts molecules by size, we will be able to read the sequence from the gel.

When we use fluorescent chain terminators, we can actually run the products of all four reactions in a single lane on a gel. The image below shows a large number of such lanes, which are illuminated with a UV laser for automated reading.

In the course of the human genome project, many new technologies were developed that greatly reduced the cost of DNA sequencing. The figure below shows a semilog plot of the cost of sequencing a human genome from the completion of the Human Genome Project in 2001 to the present day. The white line along the top is Moore's Law, the doubling of transistor density and many other measures of computing power every year. The cost of sequencing an individual human genome shows a sharp break at the end of 2007, when several new sequencing technologies were introduced.
